
Como é o cocô de quem tem fibrose cística?
Para entender melhor a fibrose cística: Quando atinge o trato intestinal pode haver produção de fezes volumosas, gordurosas e com mal cheiro ou prisão de ventre, por exemplo.
Tosse que não passa, suor mais salgado que o normal, pneumonia de repetição, diarreia e dificuldade para ganhar peso e estatura. Se esses sintomas aparecerem com frequência, pode indicar fibrose cística.
O Teste do Pezinho é a primeira triagem de Fibrose Cística na infância. Quando positivo, deve ser repetido e confirmado pelo Teste Genético ou pelo Teste do Suor. Quando o nível de cloro é superior a 60 milimoles por litro em duas dosagens, associado a um quadro clínico característico, o diagnóstico é firmado.
Nas últimas décadas, diversos avanços no diagnóstico e tratamento da FC mudaram o cenário da doença, com aumento expressivo da expectativa de vida. De acordo com dados da Cystic Fibrosis Foundation Patient Registry, a mediana da sobrevida nos EUA é de 46,2 anos,(5) sendo que no Brasil essa estimativa é de 43,8 anos.
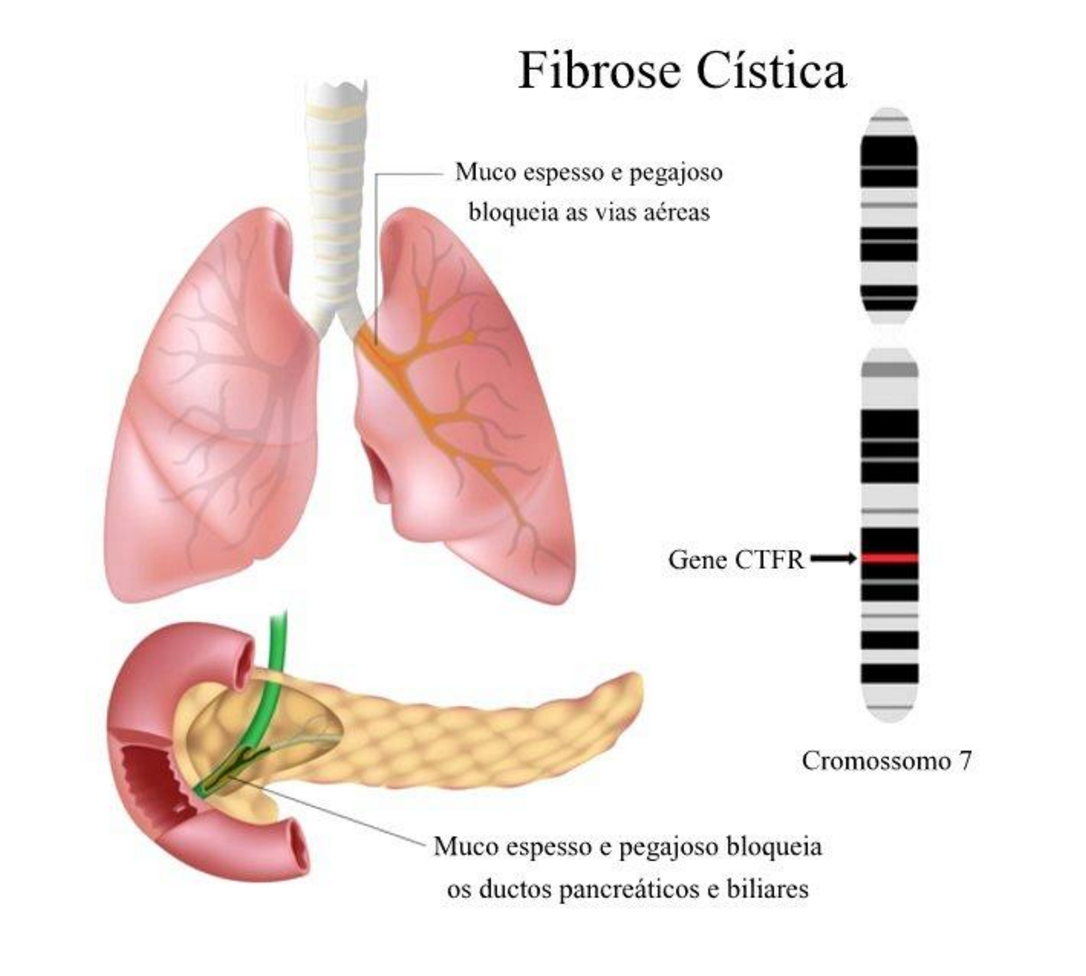
O muco espesso leva ao acúmulo de bactéria e germes nas vias respiratórias, podendo causar inchaço, inflamações e infecções como pneumonia e bronquite, trazendo danos aos pulmões. Esse muco também pode bloquear o trato digestório e o pâncreas, o que impede que enzimas digestivas cheguem ao intestino.